Medicine, Dentistry, and Pharmacy
January 18, 2024
Progression of ALS linked to a membrane and an enzyme: mitochondria-endoplasmic reticulum disruption and loss of TBK1 activity
Researchers at Nagoya University in Japan have discovered a relationship between the progression of amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, and the disruption of mitochondria-associated membranes (MAM), the contact point between the mitochondria and the endoplasmic reticulum (ER) of the cell. This discovery, published in the Proceedings of the National Academy of Sciences, provides important information about the mechanisms behind this neurodegenerative condition.
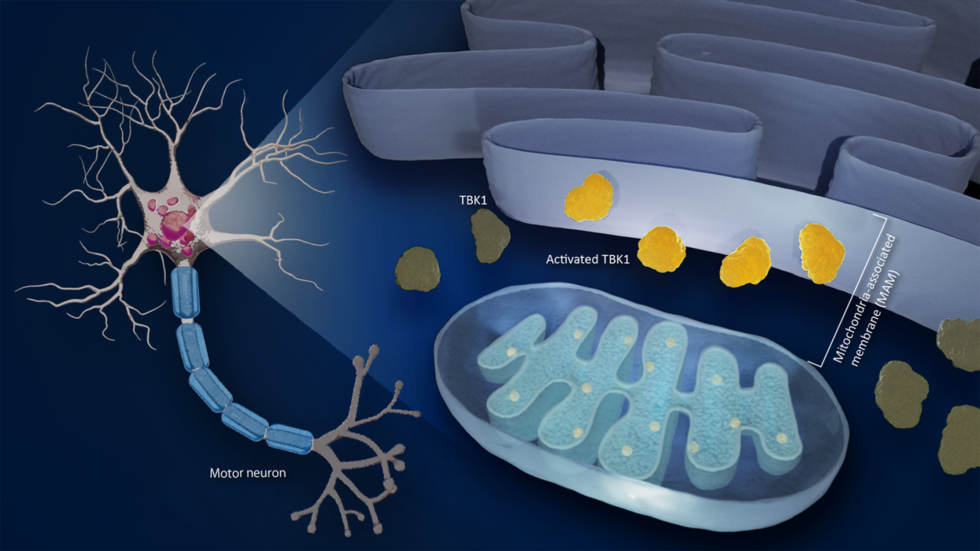
ALS is a complex disease that affects motor neurons. Previous studies have found that mitochondria, the energy-generating cells of the body, and the ER, a complex membrane network that serves protein synthesis, metabolism, and calcium storage, are involved. The MAM is where the ER interfaces with mitochondria. Although both mitochondria and ER abnormality, especially at the MAM, play a role in the progression of the disease, it is not clear how.
One possible agent is TANK-binding kinase 1 (TBK1), an enzyme that plays a crucial role in various biological processes, including inflammation and clearing damaged proteins from cells. Its disruption is important in the development of many diseases. Although mutations in the TBK1 gene cause ALS, it is not clear how TBK1 malfunctions lead to its development.
A team led by Koji Yamanaka at Nagoya University’s Research Institute of Environmental Medicine, in collaboration with Masahisa Katsuno of the Graduate School of Medicine discovered that brain and spinal cord tissues in ALS patients as well as mice with a disrupted MAM showed decreased activation of TBK1.
Seiji Watanabe, the first author of the study, explained: "TBK1 is crucial for stress response in motor neurons. If we reduce its activity, it will result in reduced tolerance to stressors, leading to neurotoxicity and eventual motor neuron death. This finding is particularly significant because abnormal protein accumulation and the resulting stress cause ALS and other neurodegenerative diseases."
When MAM is disrupted in ALS, TBK1 activity decreases. When the researchers administered arsenite, an agent that lowers TBK1 activity and disrupts MAM, to mice, they found that the mice exhibited motor problems similar to ALS.
Yamanaka added, "Our study strongly suggests that MAM significantly influences the stress response of motor neurons through TBK1 activation. Our study is consistent with human genetic studies that reported that TBK1 mutations cause ALS. Restoring TBK1 activity emerges as a potential therapeutic strategy to counter ALS, marking a promising direction for future research endeavors."
By focusing on the TBK1 pathway, the researchers have found a critical foundation for developing new ways to treat ALS and possibly other brain disorders. “We expect these results to lead to the development of new therapeutic strategies for ALS in the future,” said Yamanaka. “In future projects, restoring the TBK1 activity (or function) may be a therapeutic strategy to treat ALS.”
The study, “Mitochondria-associated membrane collapse impairs TBK1-mediated proteostatic stress response in ALS,” was published in the Proceedings of the National Academy of Sciences on November 15, 2023, at DOI: 10.1073/pnas.2315347120.
Authors:
Seiji Watanabe, Yuri Murata, Yasuyoshi Oka, Kotaro Oiwa, Mai Horiuchi, Yohei Iguchi, Okiru Komine, Akira Sobue, Masahisa Katsuno, Tomoo Ogi, Koji Yamanaka
Media Contact:
Matthew Coslett
International Communications Office, Nagoya University
Email: icomm_research@t.mail.nagoya-u.ac.jp
Top image: Research revealed that diminished activities of an enzyme called TBK1 in mitochondrial-associated membrane (MAM) reduces motor neurons’ tolerance to stressors, which is known to be causative factors in ALS. (credit:Reiko Matsushita)

