・皮膚がんや神経症状を発症する遺伝性難病である、色素性乾皮症 (xeroderma pigmentosum: XP; 指定難病159)の新たな相補性群 XP-Jと責任遺伝子GTF2H4/XPJを同定した。
・日本人のXP発症率は2〜3万人に1人 (国内の患者数は500人ほどと推定されている)で、遺伝性疾患の中では患者数が多い。
・XP-Jは、GTF2H4/XPJ遺伝子の異常により発症することを明らかにした。
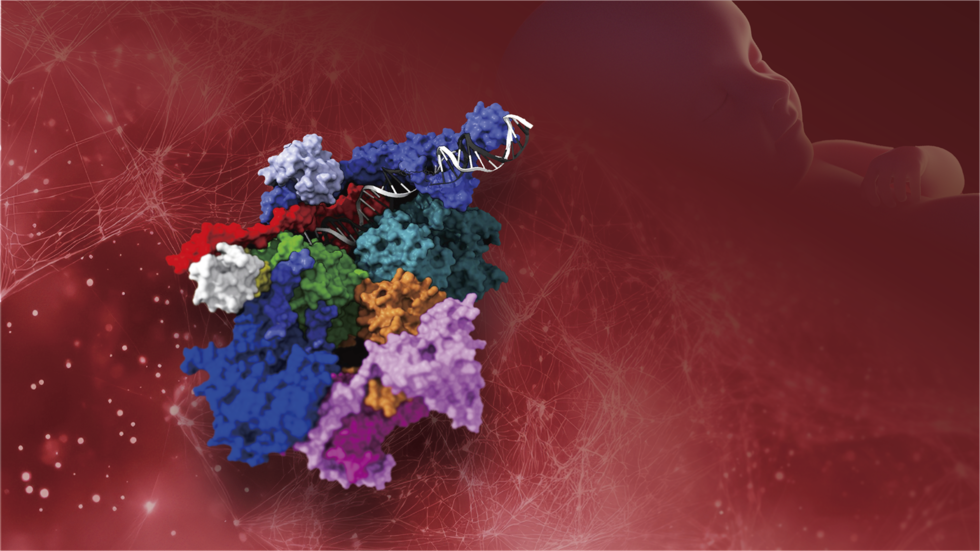
・GTF2H4/XPJ遺伝子は、ヌクレオチド除去DNA修復 (NER)に関与するTFIIH複合体 (基本転写因子複合体)の構成因子の1つp52蛋白質をコードしており、さらなる研究によりXPや関連疾患の病態解明につながると期待される。
名古屋大学大学院医学系研究科附属神経疾患・腫瘍分子医学研究センター 分子遺伝学の中沢 由華 教授、環境医学研究所 発生遺伝分野/難病ゲノム解析センターの荻 朋男 教授らの研究グループは、Rare Disease UK (英国)のHiva Fassihi医師、Shehla Mohammed医師、Heather Faucett 技師、Alan Lehmann教授らとの共同研究で、指定難病の1つである色素性乾皮症 (xeroderma pigmentosum: XP)の新たな相補性群XP-Jと責任遺伝子GTF2H4/XPJを特定しました。
XPは、生まれつきDNA損傷を修復する機構に障害があるため、紫外線や体内で生成される化学物質によって生じたDNA損傷が適切に修復されず、その結果、日光が当たる部位に皮膚がんを生じやすく、進行性の神経障害も高頻度に認められる疾患です。日本、アメリカ、ヨーロッパなどで比較的多くの患者が報告されています。
これまでに、XP-A〜G群およびXP-Vの8つの相補性群が知られており、XP-J群は50年ぶりに特定された9番目の相補性群になります。症例の解析から、XP-J群は、GTF2H4/XPJ遺伝子の異常により発症することを明らかにしました。GTF2H4/XPJ遺伝子は、ヌクレオチド除去修復機構 (nucleotide excision repair: NER)に関与するTFIIH複合体 (基本転写因子複合体)の構成蛋白質p52をコードしています。TFIIH複合体構成因子の先天的な異常では、XPのほか、早期老化を示すコケイン症候群 (Cockayne syndrome: CS)や発育発達異常を示すトリコチオジストロフィー (Trichothiodystrophy: TTD)なども発症することが知られています。本研究は、これらの発症メカニズムや分子病態の解明につながると期待されます。
本研究成果は、2025年9月9日12:00 PM EDT(日本時間9月10日01:00 AM)に国際学術誌『Journal of Clinical Investigation (JCI)』のオンライン版に掲載されました。また、2025年10月発行の印刷版に掲載予定です。
◆詳細(プレスリリース本文)はこちら
◆詳細(プレスリリース英文)はこちら
雑誌名:Journal of Clinical Investigation
論文タイトル:XP-J, a ninth xeroderma pigmentosum complementation group, results from mutations in GTF2H4, encoding TFIIH-p52 subunit.
著者:Hiva Fassihi, Shehla Mohammed, Yuka Nakazawa (名大分子遺伝), Heather Fawcett, Sally Turner, Joanne Palfrey, Isabel Garrood, Adesoji Abiona, Ana Morley, Mayuko Shimada (名大環研/難病ゲノムセンター), Kana Kato (名大環研/難病ゲノムセンター), Alan Lehmann, Tomoo Ogi (名大環研/難病ゲノムセンター)
DOI: 10.1172/JCI195731